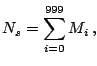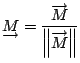Using the information from the Delaunay tessellation of a protein's
backbone, it is possible to build a statistical representation of that
protein, which takes into account the way its sequence must ''twist
and turn'' in order to bring each four-body residue cluster into
contact. Each residue - ![]() , and
, and ![]() of a four-body cluster
comprising a simplex are nearest neighbors in Euclidean space as
defined by the tessellation, but are separated by the three
distances -
of a four-body cluster
comprising a simplex are nearest neighbors in Euclidean space as
defined by the tessellation, but are separated by the three
distances -
![]() , and
, and ![]() in sequence space. Based
on this idea, we build a
in sequence space. Based
on this idea, we build a ![]() -tuple representation of a single protein
by making use of two metrics: (1) the Euclidean metric used to define
the Delaunay tessellation of the protein's C
-tuple representation of a single protein
by making use of two metrics: (1) the Euclidean metric used to define
the Delaunay tessellation of the protein's C
![]() atomic
coordinates and (2) the distance between residues in sequence space.
atomic
coordinates and (2) the distance between residues in sequence space.
If we consider a tessellated protein with N residues integrally
enumerated according to their position along the primary sequence, the
length of a simplex edge in sequence space can be defined as
![]() , where
, where ![]() is the length of the simplex edge,
is the length of the simplex edge,
![]() , corresponding to the
, corresponding to the ![]() th and
th and ![]() th
th ![]() -carbons along the sequence. If one considers the graph formed by the
union of the simplex edge between the two points
-carbons along the sequence. If one considers the graph formed by the
union of the simplex edge between the two points ![]() and
and ![]() and the
set of edges between all
and the
set of edges between all ![]() points along the sequence between
points along the sequence between
![]() and
and ![]() , it is seen that the Euclidean simplex edge,
, it is seen that the Euclidean simplex edge,
![]() , can generally be classified as a far edge (Pandit
and Amritkar, 1999). Every simplex in the protein's tessellation will
have three such edges associated with its vertices:
, can generally be classified as a far edge (Pandit
and Amritkar, 1999). Every simplex in the protein's tessellation will
have three such edges associated with its vertices: ![]() ,
, ![]() ,
, ![]() , and
, and
![]() where
where ![]() , and
, and ![]() are integers corresponding to C
are integers corresponding to C
![]() atoms enumerated according to their position along the primary
sequence. Thus, we proceed to quantify the degree of ''farness'' in
an intuitive way, by applying a transformation,
atoms enumerated according to their position along the primary
sequence. Thus, we proceed to quantify the degree of ''farness'' in
an intuitive way, by applying a transformation, ![]() , which maps the
length,
, which maps the
length, ![]() , of each edge to an integer value according to
, of each edge to an integer value according to
Condition 1 is provided because simplices with a Euclidean edge length
above
![]() are generally a result of the positions of
are generally a result of the positions of
![]() -carbons on the exterior of the protein. We filter out
contributions from these simplices to
-carbons on the exterior of the protein. We filter out
contributions from these simplices to ![]() , because they do not
represent physical interactions between the participating
residues. The simplices with the long edges are formed due to the
absence of solvent and other molecules around the protein in the
tessellation, they would not have existed if the protein was
solvated. The data structure,
, because they do not
represent physical interactions between the participating
residues. The simplices with the long edges are formed due to the
absence of solvent and other molecules around the protein in the
tessellation, they would not have existed if the protein was
solvated. The data structure, ![]() , contains
, contains
![]() elements. The number of elements is invariant with
respect to the number of residues of the protein. In order to more
easily conceptualize the mapping of the protein topology to the data
structure,
elements. The number of elements is invariant with
respect to the number of residues of the protein. In order to more
easily conceptualize the mapping of the protein topology to the data
structure, ![]() , we rewrite it as a
, we rewrite it as a ![]() -tuple vector
-tuple vector
![]() .
.
Given that each element of this vector represents a statistical
contribution to the global topology, a comparison of two proteins
making use of this mapping must involve the evaluation of the
differences in single corresponding elements of the proteins'
![]() -tuples. We define, therefore, a raw topological score,
-tuples. We define, therefore, a raw topological score, ![]() ,
representative of the topological distance between any two proteins
represented by data structures,
,
representative of the topological distance between any two proteins
represented by data structures,
![]() and
and
![]() , as the supremum norm,
, as the supremum norm,
This topological score has an obvious dependence on the sequence length difference between the two proteins being compared due to the following implicit relation for a single protein representation,
The results of topological protein structure comparison can be
illustrated using an example of proteins that belong to the same
family. Six protein families were selected from the FSSP (Families of
Structurally Similar Proteins) database for topological evaluation. We
selected families that span various levels of secondary structural
content. The representatives of these families are as follows: 1alv
and 1avm (having greater than ![]()
![]() -helical content),
2bbk and 2bpa (having greater than
-helical content),
2bbk and 2bpa (having greater than ![]()
![]() -sheet
content), and 1hfc and 1plc (having at least
-sheet
content), and 1hfc and 1plc (having at least ![]() content that
is classified as neither
content that
is classified as neither ![]() -helical nor
-helical nor ![]() -sheet). The
FSSP database contains the results of the alignments of the extended
family of each of these representative chains. Each family in the
database consists of all structural neighbors excluding very close
homologs (proteins having a sequence identity greater than
-sheet). The
FSSP database contains the results of the alignments of the extended
family of each of these representative chains. Each family in the
database consists of all structural neighbors excluding very close
homologs (proteins having a sequence identity greater than
![]() ). The topological score was calculated for each
representative in a one-against-all comparison with its neighbors. All
of the scores are plotted against RMSD for each of the families in
Fig. 3.9. A strong correlation between the topological score
and structure similarity and the power-law trend can be seen for all
families.
). The topological score was calculated for each
representative in a one-against-all comparison with its neighbors. All
of the scores are plotted against RMSD for each of the families in
Fig. 3.9. A strong correlation between the topological score
and structure similarity and the power-law trend can be seen for all
families.
![$\displaystyle T:d \mapsto \left\{ \begin{array}{ll} 1&\;\text{if}\;d=0 \\ [-.4m...
...ext{if}\;50\le d\le 100 \\ [-.4mm] 10&\;\text{if}\;d\ge 101 \end{array} \right.$](img8182.gif)
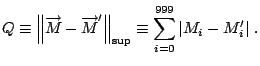
![\includegraphics[width=117mm,clip]{text/4-3/fig9.eps}](img8194.gif)