Delaunay simplices obtained as a result of the tessellation can be used to define objectively the nearest neighbor residues in 3D protein structures. The most significant feature of Delaunay tessellation, as compared with other methods of nearest neighbor identification, is that the number of nearest neighbors in three dimensions is always four, which represents a fundamental topological property of 3D space. Statistical analysis of the amino acid composition of Delaunay simplices provides information about spatial propensities of all quadruplets of amino acid residues clustered together in folded protein structures. The compositional statistics can be also used to construct four-body empirical contact potentials, which may provide improvement over traditional pairwise statistical potentials (e.g., Miyazawa and Jernigan, 2000) for protein structure analysis and prediction.
To perform the tessellation protein residues should be represented by
single points located, for example, in the positions of the
C![]() atoms or the centers of the side chains. Tessellation
training set includes high-quality representative protein structures
with low primary-sequence identity (Wang and Dunbrack, 2003). The
tessellated proteins are analyzed by computing various geometrical
properties and compositional statistics of Delaunay simplices.
atoms or the centers of the side chains. Tessellation
training set includes high-quality representative protein structures
with low primary-sequence identity (Wang and Dunbrack, 2003). The
tessellated proteins are analyzed by computing various geometrical
properties and compositional statistics of Delaunay simplices.
An example of Delaunay tessellation of a folded protein is illustrated
on Fig. 3.3 for crambin (
![]() crn). The tessellation of
this
crn). The tessellation of
this ![]() -residue protein generates an aggregate of
-residue protein generates an aggregate of ![]() nonoverlapping,
space-filling irregular tetrahedra (Delaunay simplices). Each Delaunay
simplex uniquely defines four nearest neighbor C
nonoverlapping,
space-filling irregular tetrahedra (Delaunay simplices). Each Delaunay
simplex uniquely defines four nearest neighbor C
![]() atoms and
thus four nearest neighbor amino acid residues.
atoms and
thus four nearest neighbor amino acid residues.
For the analysis of correlations between the structure and sequence of
proteins, we introduced a classification of simplices based on the
relative positions of vertex residues in the primary sequence (Singh
et al., 1996). Two residues were defined as distant if they were
separated by one or more residues in the protein primary
sequence. Simplices were divided into five nonredundant classes: class
![]() , where all four residues in the simplex are consecutive in the
protein primary sequence; class
, where all four residues in the simplex are consecutive in the
protein primary sequence; class ![]() , where three residues are
consecutive and the fourth is a distant one; class
, where three residues are
consecutive and the fourth is a distant one; class ![]() , where
two pairs of consecutive residues are separated in the sequence; class
, where
two pairs of consecutive residues are separated in the sequence; class
![]() , where two residues are consecutive, and the other two are
distant both from the first two and from each other; and class
, where two residues are consecutive, and the other two are
distant both from the first two and from each other; and class
![]() where all four residues are distant from each other
(Fig. 3.4). All five classes usually occur in any given
protein.
where all four residues are distant from each other
(Fig. 3.4). All five classes usually occur in any given
protein.
The differences between classes of simplices can be evaluated using
geometrical parameters of tetrahedra such as volume and tetrahedrality
(3.1). Distributions of volume and tetrahedrality for all five
classes of simplices is shown in Fig. 3.5. The sharp narrow
peaks correspond to the simplices of classes ![]() and
and
![]() . They tend to have well defined distributions of volume and
distortion of tetrahedrality. These results suggest that tetrahedra of
these two classes may occur in regular protein conformations such as
. They tend to have well defined distributions of volume and
distortion of tetrahedrality. These results suggest that tetrahedra of
these two classes may occur in regular protein conformations such as
![]() -helices and may be indicative of a protein fold family. We
have calculated the relative frequency of occurrence of tetrahedra of
each class in each protein in a small dataset of hundred proteins from
different families and plotted the results in Fig. 3.6. The
proteins were sorted in the ascending order of fraction of tetrahedra
of class
-helices and may be indicative of a protein fold family. We
have calculated the relative frequency of occurrence of tetrahedra of
each class in each protein in a small dataset of hundred proteins from
different families and plotted the results in Fig. 3.6. The
proteins were sorted in the ascending order of fraction of tetrahedra
of class ![]() . Noticeably, the content of simplices of class
. Noticeably, the content of simplices of class
![]() decreases with the increase of the content of class
decreases with the increase of the content of class ![]() simplices. According to common classifications of protein fold
families (Orengo et al., 1997), at the top level of hierarchy most
proteins can be characterized as all-alpha, all-beta, or
alpha/beta. The fold families for the proteins in the dataset are also
shown in Fig. 3.6. These results suggest that proteins having
a high content of tetrahedra of classes
simplices. According to common classifications of protein fold
families (Orengo et al., 1997), at the top level of hierarchy most
proteins can be characterized as all-alpha, all-beta, or
alpha/beta. The fold families for the proteins in the dataset are also
shown in Fig. 3.6. These results suggest that proteins having
a high content of tetrahedra of classes ![]() and
and ![]() (i.e.,
proteins in the right part of the plot in Fig. 3.6) belong to
the family of all-alpha proteins. Similarly, proteins having a low
content of tetrahedra of classes
(i.e.,
proteins in the right part of the plot in Fig. 3.6) belong to
the family of all-alpha proteins. Similarly, proteins having a low
content of tetrahedra of classes ![]() and
and ![]() but a high
content of tetrahedra of classes
but a high
content of tetrahedra of classes ![]() and
and ![]() (i.e.,
proteins in the left part of the plot in Fig. 3.6) belong to
the all-beta protein fold family. Finally, proteins in the middle of
the plot belong to the alpha/beta fold family. Thus, the results of
this analysis show that the ratio of tetrahedra of different classes
is indicative of the protein fold family.
(i.e.,
proteins in the left part of the plot in Fig. 3.6) belong to
the all-beta protein fold family. Finally, proteins in the middle of
the plot belong to the alpha/beta fold family. Thus, the results of
this analysis show that the ratio of tetrahedra of different classes
is indicative of the protein fold family.
![\includegraphics[width=117mm,clip]{text/4-3/fig6.eps}](img8157.gif)
|
Identification of significant patterns in biomolecular objects depends
on the possibility to distinguish what is likely from what is unlikely
to occur by chance (Karlin et al., 1991). Statistical analysis of
amino acid composition of the Delaunay simplices provides information
about spatial propensities of all quadruplets of amino acid residues
to be clustered together in folded protein structures. We analyzed the
results of the Delaunay tessellation of these proteins in terms of
statistical likelihood of occurrence of four nearest neighbor amino
acid residues for all observed quadruplet combinations of ![]() natural
amino acids. The log-likelihood factor,
natural
amino acids. The log-likelihood factor, ![]() , for each quadruplet was
calculated using the following equation:
, for each quadruplet was
calculated using the following equation:
Theoretically, the maximum number of all possible quadruplets of
![]() natural amino acid residues is
natural amino acid residues is ![]()
![]() . The first term accounts
for simplices with four distinct residue types, the second - three
types in
. The first term accounts
for simplices with four distinct residue types, the second - three
types in ![]() distribution, the third - two types in
distribution, the third - two types in ![]() distribution, the fourth - two types in
distribution, the fourth - two types in ![]() distribution, and the
fifth - four identical residues. The log-likelihood factor
distribution, and the
fifth - four identical residues. The log-likelihood factor ![]() is
plotted in Fig. 3.7 for all observed quadruplets of natural
amino acids. Each quadruplet is thus characterized by a certain value
of the
is
plotted in Fig. 3.7 for all observed quadruplets of natural
amino acids. Each quadruplet is thus characterized by a certain value
of the ![]() factor which describes the nonrandom bias for the four
amino acid residues to be found in the same Delaunay simplex. This
value can be interpreted as a four-body statistical potential energy
function. The statistical potential can be used in a variety of
structure prediction, protein modeling, and computational mutagenesis
applications.
factor which describes the nonrandom bias for the four
amino acid residues to be found in the same Delaunay simplex. This
value can be interpreted as a four-body statistical potential energy
function. The statistical potential can be used in a variety of
structure prediction, protein modeling, and computational mutagenesis
applications.
Computational mutagenesis is based on the analysis of a protein potential profile, which is constructed by summing the log-likelihood scores from (3.2) for all simplices in which a particular residue participates. A plot of the potential profile for a small protein, HIV-1 protease, is shown in Fig. 3.8. The shape of the potential profile frequently reflects important features of the protein, for example, the residues in local maxima values of the profile are usually located in the hydrophobic core of the protein and these residues play an important role in maintaining protein stability.
A potential profile can be easily calculated for both wild type and mutant proteins, assuming that the structural differences between them are small and that their tessellation results are similar. In this case the difference between the profiles is defined only by the change in composition of the simplices involving the substitution site. The resulting difference profile provides important insights into the changes in protein energetics due to the mutation.
![\includegraphics[width=55mm,clip]{text/4-3/fig3.eps}](img8152.gif)
![\includegraphics[clip]{text/4-3/fig4.eps}](img8155.gif)
![\includegraphics[width=100mm,clip]{text/4-3/fig5.eps}](img8156.gif)
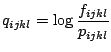
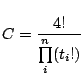
![\includegraphics[width=105mm,clip]{text/4-3/fig7.eps}](img8174.gif)
![\includegraphics[width=108mm,clip]{text/4-3/fig8.eps}](img8175.gif)